エクソーム解析
特長
- ゲノムのエクソン領域のみを濃縮してシーケンシングするため、シーケンスコストを大幅に低減。
- Single Nucleotide Variation(SNV)、Short Insertion/Deletion(InDel)の探索と弊社が独自開発したシステムによるアノテーション付与。
- 遺伝性疾患からがん※まで候補遺伝子の同定に有用。
- ※ 体細胞変異の解析をご希望の場合は、こちらのページをご参照下さい。
理研ジェネシス Whole Exome Sequencingの強み
- 数万検体規模の調査・コホート研究に対応。
- 国内トップレベルの解析実績・大規模プロジェクトでの採用実績。
- ゲノム解析の経験豊富なスタッフによる安心のサービスとサポート体制。
- 医薬品開発に対応した品質保証体制での実施。
解析概要

エクソン領域濃縮キット
SureSelect All Exon
実績のあるAgilent technologies社のSureSelect All Exonを使用したサービスです。| パネル名 | 対応生物種 | 対象領域 | デザイン サイズ |
|---|---|---|---|
| SureSelect Human All Exon V6 | ヒト | コーディング領域、miRNA、snoRNA、tRNA | 60 Mb |
| SureSelect Human All Exon V6 + UTR | ヒト | V6の全対象領域、体質性疾患研究向けUTR領域 | 91 Mb |
| SureSelect Human All Exon V7 | ヒト | コーディング領域 | 48.2 Mb |
| SureSelect Human All Exon V8 | ヒト | コーディング領域、TERTプロモーター | 41.6 Mb |
| SureSelect Mouse All Exon | マウス | コーディング領域 | 49.6 Mb |
Twist Exome
Twist Bioscience社のエクソーム用パネルを使用したサービスです。最適化された高品質の二本鎖DNAプローブを使用することで、より均一なカバレッジを持つデータが取得でき、シーケンス効率の向上とコストの削減が可能になりました。| パネル名 | 対応生物種 | 対象領域 | ターゲット サイズ |
|---|---|---|---|
| Human Core Exome | ヒト | コーディング領域(CCDSのコーディング領域を99%以上カバー) | 33.2 Mb |
| Human Core Exome + RefSeq またはComprehensive Exome |
ヒト | コーディング領域(CCDS、RefSeq、GENCODEのコーディング領域を99%以上カバー) | 36.8 Mb |
解析結果
検出されたSNVは、各種アノテーション※2をつけてエクセル・マクロファイル※3 とテキストファイルでご提出します。エクセル・マクロファイル上で、Compound Heterozygote(複合ヘテロ接合体)変異候補を抽出することも可能です(この機能をご使用いただくためには、トリオサンプルのデータが必要です)。
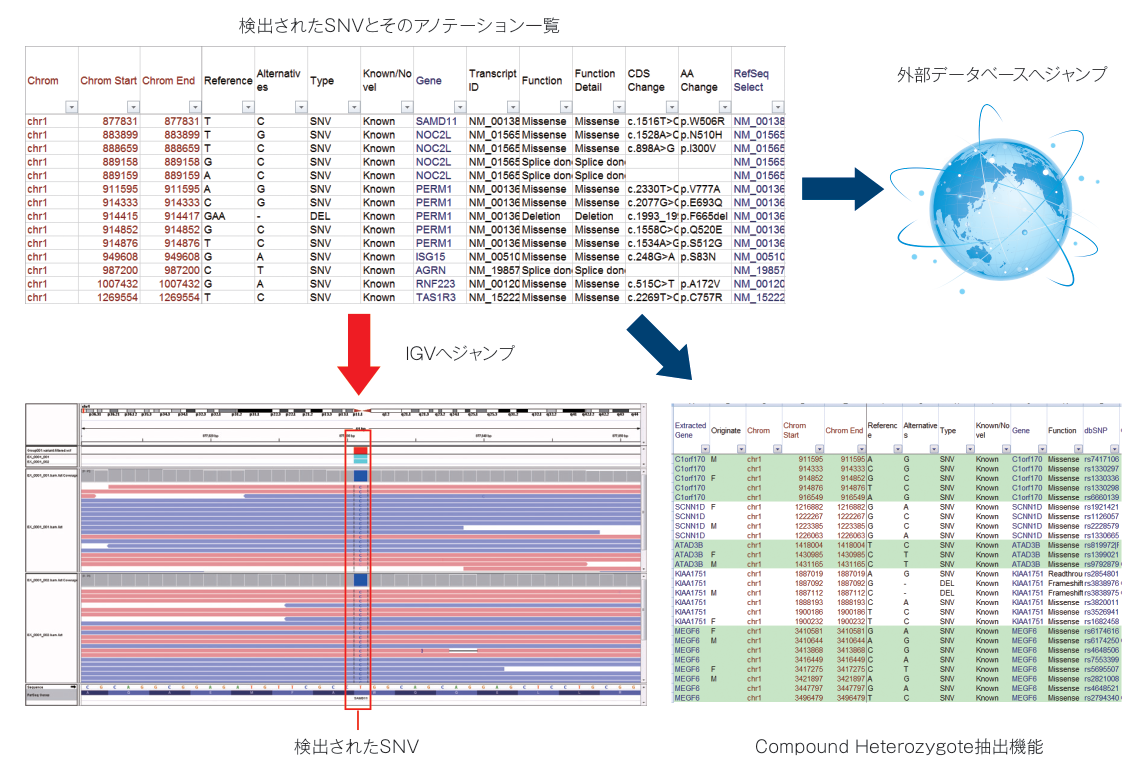
- ※1 マッピング結果、SNV、DepthはIntegrative Genomics Viewer(IGV)で閲覧することが可能です。
- ※2 検出されたSNVには次のようなアノテーションがつきます(Humanの場合)。
変異箇所、塩基、新規/既知、影響を与える遺伝子、遺伝子への影響、dbSNP ID、RefSeq ID、CCDS ID、アレル頻度(gnomAD、1000 Genomes、HGVD、GEM-J WGA)、Conserved Elements、アミノ酸置換影響予測(SIFTなど)、Splice Site変異影響予測、Depth、クオリティ値、サンプルごとのジェノタイプデータなど。 - ※3 アノテーションは弊社オリジナルのエクセル・マクロ(AnnotationViewer)で観覧でき、IGVおよび外部データベースと連携するようになっています。
納品物
- 解析報告書
- データHDD:リード情報(FASTQ)、マッピングデータ(BAM)、SNV/InDel検出データ(VCF)、SNV/InDelのアノテーションデータ(エクセル・マクロファイルとテキストファイル)
サンプル条件
| エクソン領域濃縮キット | SureSelect All Exon | Twist Exome |
|---|---|---|
| サンプルの種類 | 精製ゲノムDNA | 精製ゲノムDNA |
| DNA量 | 3 µg | 0.3 µg |
| 濃度 | 50 ng/µL以上 | 10 ng/µL以上 |
| OD260/280 | 1.8以上 | 1.8以上 |
| 溶媒 | 1x TE | 10 mM Tris-HCl (pH 8.0) またはNuclease-free水※ |
- ※ EDTAを含まないバッファーを溶媒としてご使用ください。
- サンプルの定量はQubitまたはPicoGreenを用いた方法を推奨しております。
- サンプルの電気泳動写真がお手元にある場合にはご提供をお願いします。
解析例
| 機種 | NovaSeq 6000 |
|---|---|
| Exon Capture方法 | SureSelect Human All Exon V6 |
| 平均データ量/サンプル | 約10 Gb |
| リード長 | 150 bp |
| シーケンス方法 | Paired End/Multiplex |
| バイオインフォマティクス解析 | SNVおよび一部のShort InDelの同定、 各種アノテーション付与 |
| 納期 | 品質評価通過後、約2.5ヵ月※ |
- ※ 同時に解析を行うサンプル数が多い場合には、別途お打ち合わせの上決定いたします。