特長
解析概要
融合遺伝子は、あるタイプのがんでは顕著な特徴となっており、分子標的医薬のターゲットとして着目されています。 がん細胞などに由来する転写産物の配列を網羅的に決定した後、それらの配列情報から融合遺伝子を探索します。
解析結果
シーケンス解析により取得したペアエンドリード配列から、ソフトウェア(STAR-Fusion)を使用して、融合遺伝子を検出します。
検出された融合遺伝子の情報(Pfam domain、インフレームか否かの判定結果、breakpoint前後の配列、ゲノム位置などの詳細情報)をサマリーレポートと検出結果リストで、支持リードの状況をHTML形式のファイルでご提出します。
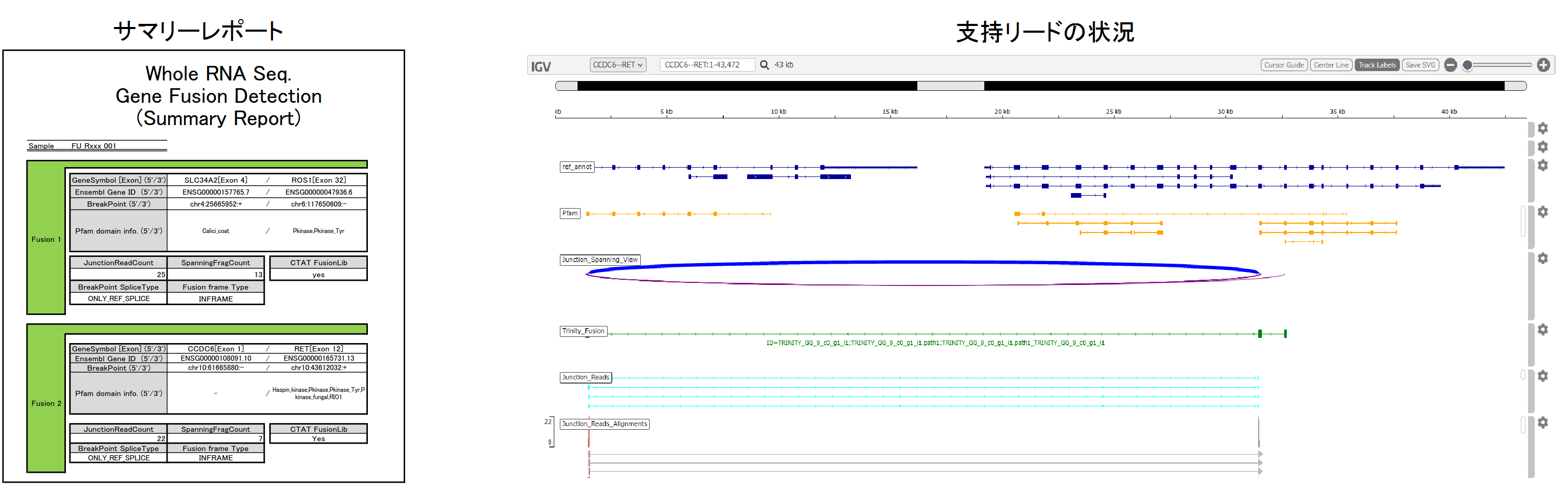
納品物
- 解析報告書
- データHDD: リード情報(FASTQ)、サマリーレポート(PDF)、検出結果リスト(Excel)、支持リード情報(HTML)
サンプル条件
| ライブラリ作製キット |
TruSeq Stranded mRNA Library Prep |
TruSeq RNA Exome |
| サンプルの種類 |
精製Total RNA |
精製Total RNA |
| RNA量 |
3 µg |
0.5 µg |
| 濃度※1 |
65 ng/µL以上 |
20 ng/µL以上 |
| RIN値※2 |
7 以上 |
-※3 |
- ※1 サンプルの定量はAgilent 2100バイオアナライザまたはAgilent 2200 TapeStationを用いた方法を推奨しております。
- ※2 サンプルの(バイオアナライザによる)電気泳動図がお手元にある場合にはご提供をお願いします。
- ※3 200 nt以上のRNA断片が50%以上(DV200≥50%)の品質を推奨しています。
解析例
| ライブラリ作製キット |
TruSeq Stranded mRNA Library Prep |
| 機種 |
NovaSeq 6000 |
| 平均リード数/サンプル |
約100M リード以上 |
| シーケンス方法 |
Paired End/Multiplex |
| バイオインフォマティクス解析 |
融合遺伝子の探索 |
| 納期 |
品質評価通過後、約3ヵ月※ |
- ※ 同時に解析を行うサンプル数が多い場合には、別途お打ち合わせの上決定いたします。