RNA sequencing (RNA-seq)
特長
理研ジェネシス RNA Sequencingの強み
- 数百検体規模の調査・コホート研究に対応。
- 様々な機関から認められた品質保証で医薬品開発に対応。
- 経験豊富なスタッフによる安心のサービスとサポート体制。
- 国内トップレベルの解析実績・大規模プロジェクトでの採用実績。
解析概要

ライブラリ作製方法
| No. | ライブラリー作製キット | RNA精製方法 | FFPE由来RNA対応 | 特徴 |
|---|---|---|---|---|
| 1 | TruSeq Stranded mRNA Library Prep※1 | poly A RNA精製 | × | 最もスタンダードな手法。分解の見られるRNAは適さない。 |
| 2 | TruSeq RNA Exome※2 | - | ○ | ヒト遺伝子(21,415)のコーディング領域のみを濃縮して解析。分解の見られるRNAでも解析可能。 |
| 3 | TruSeq Stranded Total RNA Library Prep | rRNA除去 | × | polyAを持たないタイプのnon-coding RNAもシーケンス可能。分解の見られるRNAでも解析可能。 |
| 4 | SureSelect XT RNA direct | - | ○ | マウス遺伝子のコーディング領域のみを濃縮して解析。分解の見られるRNAでも解析可能。 |
- ※1 グロビンmRNAの除去をご希望の場合は別途お問い合わせください。
- ※2 ヒトのみ対応可能。
データ解析 標準BI解析
内容
標準BIサービスでは、Illumina社DRAGEN RNA Pipelineと弊社独自のパイプラインを用いて、以下の解析を行います。
- リファレンスゲノム配列へのマッピング
- マッピングデータからのカウント行列の作成
- 発現変動遺伝子の同定とTMMカウント行列の作成
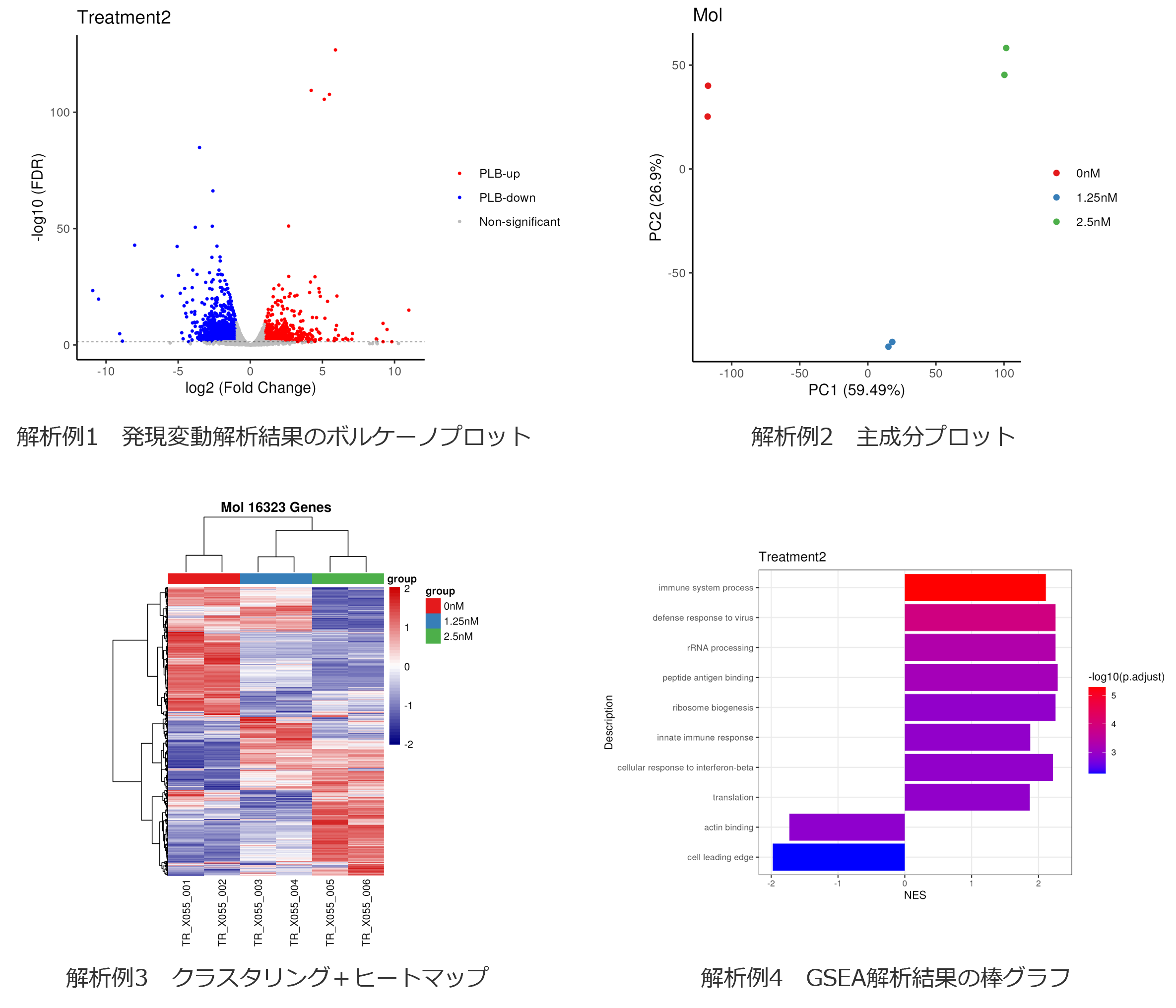
- 発現変動解析の結果の可視化(解析例1)
- 主成分分析(解析例2)
- クラスタリング結果に基づくヒートマップの作成(解析例3)
- エンリッチメント解析(二群間比較解析の場合のみ実施)(解析例4)
データ解析 カスタムBI解析
- Fusion Gene Analysis / 融合遺伝子探索(ヒトのみ対応可能)
ペアエンドリード配列を用いて、発現している融合遺伝子を効率良く探索します。
詳細はFusion Gene Analysisサービスのページをご参照ください。
サービスプラン
- 生データプラン
- 弊社でライフラリー作製・シーケンス・シーケンスデータを取得します。納品物は取得したraw data(生データ)のみを納品いたします。ご自身でデータ解析を実施される場合に適したプランです。
- 標準BI解析プラン
- 弊社でライフラリー作製・シーケンス・シーケンスデータを取得し、標準BI解析を実施するプランです。一般的な統計解析をご希望の場合に適したプランです。
- カスタムBI解析プラン
- 弊社でライフラリー作製・シーケンス・シーケンスデータを取得し、カスタムBI解析を実施するプランです。上記のカスタムBI解析メニューからご希望の解析をご選択ください。
納品物
- 解析報告書
- データHDD: リード情報(FASTQ)、マッピングデータ(BAM)、転写物データ(GTF)、データ解析結果(Excelファイルとテキストファイル)
解析条件
- 2サンプル以上※4
- ※4 ただし、統計検定の都合上、一つの群に属するサンプルレプリケート数が二以上となるようご準備いただくことを強く推奨しております。例えば、二群比較をご希望の場合には「2サンプル」×「二群」の計「4サンプル」以上ご用意いただくのが望ましいです。
サンプル条件
| ライブラリ作製キット | TruSeq Stranded mRNA Library Prep、 TruSeq Stranded Total RNA Library Prep |
TruSeq RNA Exome、 SureSelect XT RNA direct |
|---|---|---|
| サンプルの種類 | 精製Total RNA | 精製Total RNA |
| RNA量 | 3µg | 0.5µg |
| 濃度※5 | 65 ng/µL以上 | 20 ng/µL以上 |
| RIN値※6 | 7以上 | -※7 |
- ※5 サンプルの定量はAgilent 2100バイオアナライザまたはAgilent 2200 TapeStationを用いた方法を推奨しております。
- ※6 サンプルの(バイオアナライザによる)電気泳動図がお手元にある場合にはご提供をお願いします。
- ※7 200 nt以上のRNA断片が50%以上(DV200>50%)の品質を推奨しています。
解析例
| ライブラリ作製キット | TruSeq Stranded mRNA Library Prep |
|---|---|
| 機種 | NovaSeq 6000 |
| 平均データ量/サンプル | 約40M リード以上 |
| シーケンス方法 | Paired End/Multiplex |
| バイオインフォマティクス解析 | スプライシングパターン予測、正規化された発現レベルの算出、 発現レベルのサンプル間比較、GO解析 |
| 納期 | 品質評価通過後、約2.5ヵ月※8 |
- ※8 同時に解析を行うサンプル数が多い場合には、別途お打ち合わせの上決定いたします。
お問い合わせはこちら